A 7 year-old male neutered domestic shorthaired 5 kg cat is currently hospitalized for management of an aortic thromboembolism. The cat was diagnosed with hypertrophic cardiomyopathy, left sided congestive heart failure and aortic thromboembolism causing complete lack of motor function in the pelvic limbs. It is being managed with oxygen therapy provided by commercial oxygen kennel, 2 μg/kg/hr fentanyl infusion, furosemide 2 mg/kg q12 hours and 18.75 mg clopidogrel. The cat is being monitored with regular respiratory rate checks and continuous electrocardiogram (ECG). The cat began to show some improvement in pelvic limb motor function. However, 48 hours post initiation of therapy there was a dramatic change in the ECG (Figure 1).
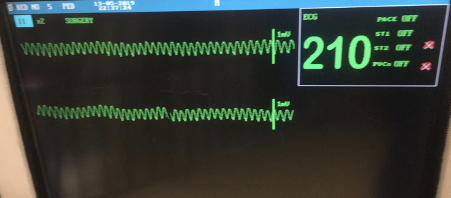
Intravenous lidocaine (0.5 mg/kg) was given immediately with no appreciable effect; a further 0.5 mg/kg lidocaine was repeated and a vagal maneuver was performed via carotid massage whilst pending results of venous blood gas, metabolite and electrolyte analysis. The venous blood gas revealed a potassium 8.7 mmol/L and creatinine 400 μmol/L (4.52 mg/dL). Therapy was initiated with 10% calcium gluconate 0.5 ml/kg, regular insulin 0.25 IU/kg and glucose 0.5 g/kg. The rhythm converted to a sinus tachycardia and the potassium reduced to 7.0 mmol/L. However, the cat was anuric and non-responsive to furosemide and therefore the owners elected to euthanize.
Now let’s consider this cat’s ECG a little further.
The ECG displays a regular repeating wide and bizarre complexes with a lack of appreciable P waves and a rate in excess of what is considered normal for a cat, in a simplest form – a wide complex tachycardia.
The most common cause of wide complex tachycardia is ventricular tachycardia (Santilli et al 2018). However, there are other important differentials (Table 1). In light of the response to therapy, hyperkalemia was considered to be the most likely cause of the wide complex tachycardia in this patient.
| Differential | ECG features |
| Ventricular tachycardia | Persistent often regular repeating wide QRS complex Atrioventricular dissociation Fusion beats +/- Capture beats |
| Supraventricular tachycardia with aberrancy Bundle branch block -Left bundle branch block -Right bundle branch block | Atrioventricular association Wide QRS left shift MEA (positive in leads I, II, avF) Wide QRS right shift MEA (negative in leads I, II, avF) |
| Atrial fibrillation with an accessory pathway | Wide QRS complex Atrioventricular association Short PR interval Delta wave |
| Sinoventricular rhythm with accelerated junctional rhythm | Absent P waves ST segment depression |
In experimental studies hyperkalemia has been shown to be most commonly been associated with bradyarrhythmias (Table 2). These include depressed P wave amplitude, prolonged PR interval, atrial standstill (absent P waves), prolongation of the QT interval, ST segment depression and peaked T waves (Ettinger et al 1974, Surawicz 1967). However, veterinary clinical studies have demonstrated more variable ECG changes secondary to hyperkalemia and tachycardia has been reported in 7-8% of hyperkalemic dogs and cats (Tag & Day 2008, Hoehne et al 2019).
| Serum K+ concentration | ECG abnormalities |
| ≥5-5-6.5mmol/L | Increased T wave amplitude |
| ≥6.6-7.0mmol/L | Decreased R wave amplitude Prolonged QRS and PR interval ST segment depression |
| ≥7.1-8.5mmol/L | Decreased P wave amplitude Increased P wave duration Prolongation of QT interval |
| ≥8.6-10.0mmol/L | Lack of P waves (atrial standstill) Sinoventricular rhythm |
| ≥10.1mmol/L | Widened and biphasic QRS complex Ventricular flutter, fibrillation or asystole |
To better understand the ECG abnormalities that can be associated with hyperkalemia we first need to understand the cardiac action potential.
There are three main types of cardiomyocytes:
- Pacemaker cells (sinoatrial and atrioventricular node, bundle of His and the purkinje system)
- Atrial myocytes
- Ventricular myocytes
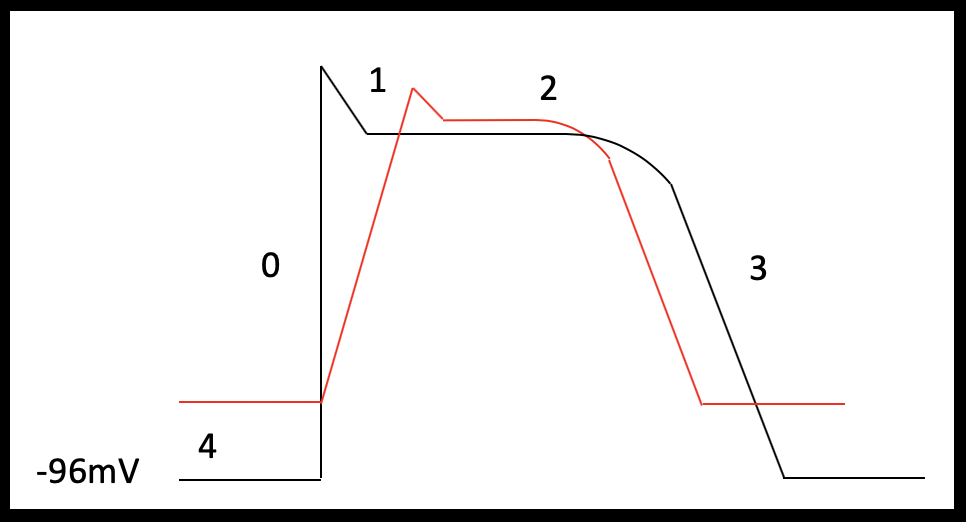
Pacemaker cells form the specialized conduction system of the heart and are therefore responsible for the intrinsic cardiac rhythm. Atrial and ventricular myocytes are capable of both electrical conduction and contraction. The shape of the cardiac action potential of atrial and ventricular myocytes are similar and can be broadly divided in to four phases (Table 3).
| Phase 0 (Fast depolarization) | Stimuli that exceeds the membrane threshold potential open voltage gated sodium channels leading to increased sodium influx and membrane depolarization. |
| Phase 1 (Partial repolarization) | Voltage gated sodium channels inactivate and fast voltage gated potassium channels open briefly. This outward potassium current leads to a brief period of repolarization. |
| Phase 2 (Plateau) | Membrane depolarization is maintained through balance of inward currents (predominantly calcium) and outward currents (slow and delayed rectifier potassium channels). Excitation-contraction coupling occurs in this phase. |
| Phase 3 (Final repolarization) | A gradual inactivation of voltage gated calcium channels reduces the inward calcium current and outward potassium currents return the membrane to its resting membrane potential. |
| Phase 4 (Resting membrane potential) | Maintained by potassium leak channels and inward rectifying potassium channels |
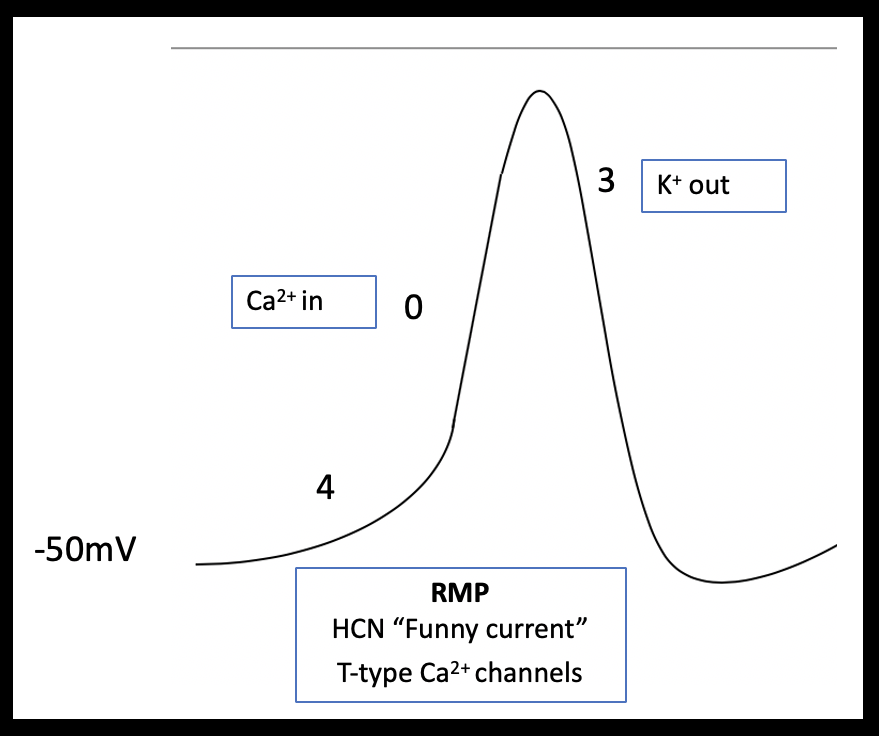
Atrial myocytes have a reduced number of calcium channels and, therefore, their more abbreviated phase 2 plateau is leading to a more triangular shaped action potential (Brandenburg et al 2016; Figure 2a and b). Pacemaker cells lack contractile function but can spontaneously depolarize. These features are demonstrated in the shape of their action potential; pacemaker cells have an unstable resting membrane potential and there is absent plateau phase. Phase 0 (fast depolarization) of pacemaker cells is predominantly calcium driven (Figure 3).
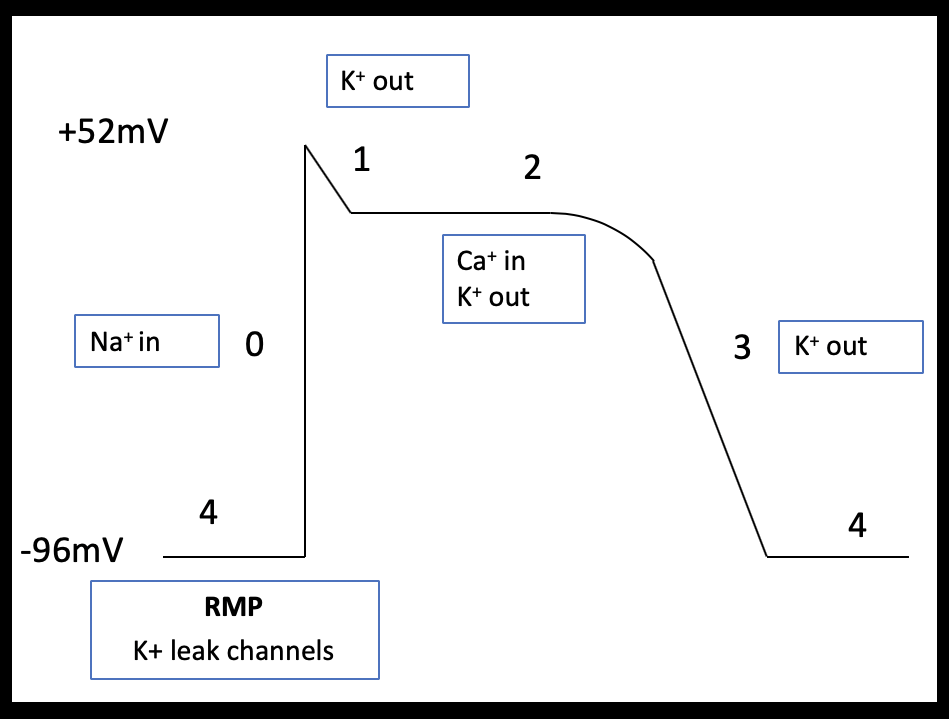
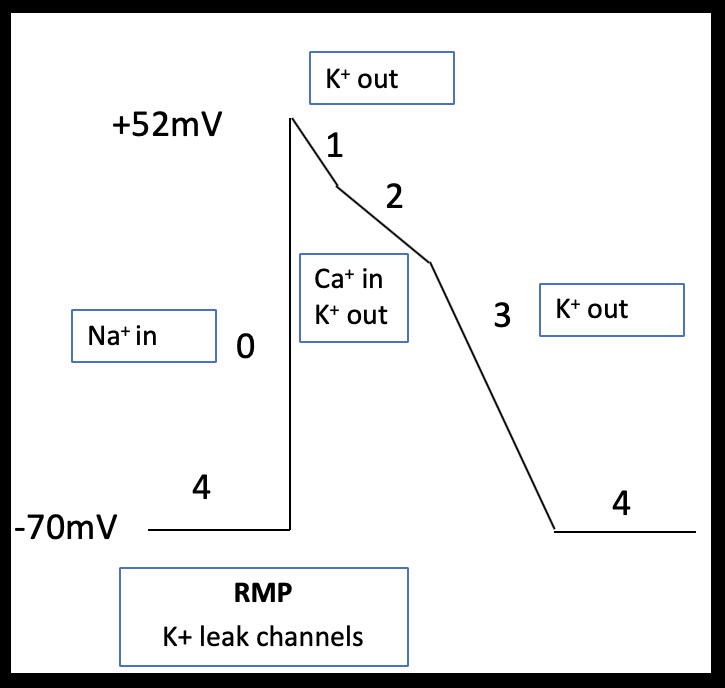
The diverse ECG secondary to hyperkalemia are dependent on the relative effect of potassium on the different phases of the action potential and any co-existing acid base or electrolyte disturbances (Tag & Day 2008)
The resting membrane potential in atrial and ventricular myocytes is maintained primarily by the concentration gradient and membrane permeability to potassium. If serum potassium levels increase, then there is a reduced concentration gradient between the intracellular and extracellular potassium concentration leading to depolarization of the membrane potential – the resting membrane potential becomes less negative. Depolarization of the membrane opens some voltage gated sodium channels and these channels are subsequently inactivated, reducing the number of sodium channels available for activation during phase 0. The overall effect of this is the reduction in the rate of rise (Vmax) of phase 0 (Vasalle et al 1964). Furthermore, for reasons not well understood, the slow and delayed potassium rectifier channels responsible for plateau phase and partial repolarization phase are sensitive to extracellular potassium levels; as potassium increases in the extracellular space, potassium conductance through these channels is increased. This leads to a shortening of repolarization (phase 2 and 3) and is thought explain the early ECG manifestations of hyperkalemia including ST segment depression, QT shortening and peaked T waves (Parham et al 2006; figure 4).
Not all cardiomyocytes have the same sensitivity to changes in potassium concentration. Pacemaker cells have been shown to be less sensitive to the effect of hyperkalemia than atrial and ventricular cardiomyocytes. The decreased sensitivity of pacemaker cells to hyperkalemia is a result of decrease in potassium conductance at the pacemaker resting membrane potential and the predominance of calcium and not sodium influx during rapid depolarization (Vasalle et al 1964). Furthermore, studies of atrial muscle have demonstrated that atrial cardiomyocytes have increased sensitivity to potassium compared to ventricular cardiomyocytes (DeMello 1960). This difference in susceptibility between the different cardiomyocytes means that even after atrial cells become unexcitable, the sinus rhythm can persist and drive the ventricles; termed sinoventricular rhythm. P waves represent atrial activity and the loss of atrial depolarization results in absent P waves and conduction through the ventricular myocardium is delayed resulting in a wide QRS morphology. The widen QRS may blend with the T wave to form a sine-wave morphology (Cohen al 1971).
Hyperkalaemia can also produce accelerated junctional rhythms and because of the ECG manifestations of hyperkalemia this appears as a wide complex tachycardia with absent P waves (McLean et al 2000). Wide complex tachycardia has been described in clinical cases of dogs and cats with hyperkalaemia (Norman et al 2006, Rubinack et al 2016). Two out of three cats and the dog in the canine case report converted to sinus tachycardia with the administration of calcium gluconate but sinus tachycardia remained until the potassium was normalized. Pain and co-existing electrolyte and mineral abnormalities such as hypocalcemia and hypomagnesemia were hypothesized to contribute to the ECG abnormalities (Norman et al 2006, Rubinack et al 2016). Hyperkalemia has also been reported to cause ventricular tachycardia as result of ventricular re-entry secondary to changes in conduction velocity. This rhythm is typically thought to immediately proceed cardiac arrest (Cohen et al 1971, Vassalle et al 1964).
Ventricular tachycardia is usually differentiated from supraventricular tachycardia by assessing for atrioventricular dissociation, and detection of fusion beats with or without capture beats (Brady & Skiles 1999). However, the absence of P waves in hyperkalemic patients makes the assessment of atrioventricular disassociation impossible and these reports highlight the difficulties in ECG interpretation of wide complex tachycardia in a hyperkalemic patient.
This is interesting physiology but the real question is: does knowing that hyperkalemia can induce a wide complex tachycardia that is not originating from the ventricles effect our decision making in a clinical setting?
A commonly used first line drug for the treatment of ventricular tachycardia is lidocaine; a class 1A sodium channel blocker. Lidocaine blocks sodium channels and exert “use dependence” preferentially binding to inactivated sodium channels. As previously discussed, hyperkalemia causes depolarization of the resting membrane potential and this depolarization inactivates a portion of sodium channels. Therefore, the action of lidocaine may be potentiated in the presence of hyperkalemia (Campbell et al 1991). If enough sodium channels are inactivated the cell membrane becomes unexcitable and this could progress to asystole. Cardiac arrest secondary to systemic lidocaine administration to a hyperkalemic patient has been described in people (McLean et al 2000, Song et al 2008) and there are anecdotal reports of cardiac arrest in hyperkalemic dogs and cats post a single dose of lidocaine. In the aforementioned canine case report the dog was non-responsive to lidocaine but no adverse effects were reported (Rubinack et al 2016).
The Bottom Line
Ventricular tachycardia is not always the answer to a wide complex tachycardia. Electrolyte analysis should be performed early in a patient with wide complex tachycardia and lidocaine administration should be carefully considered in patients previously documented to have or who are at risk of hyperkalemia.
References
Santilli R, Moïse NS, Pariaut R et al. (2018) Electrocardiography of the dog and cat, 2nd ed., Italy: Edra.
Tag TL, Day TK. Electrocardiographic assessment of hyperkalemia in dogs and cats. J Vet Emerg Crit Care 2008; 8(1): 61-67.
Hoehne S, Hopper K, Epstein SE. Retrospective evaluation of the severity of and prognosis associated with potassium abnormalities in dogs and cats presenting to an emergency room (January 2014–August 2015): 2441 cases. J Vet Emerg Crit Care 2019; 29(6): 653-661.
Ettinger PO, Regan TJ, Oldewurtel HA. Hyperkalemia, cardiac conduction, and the electrocardiogram. Am Heart J 1974; 88(3):360–369.
Surawicz B. Relationships between electrocardiogram and electrolytes. Am Heart J 1967; 73(6):814–831.
Brandenburg S., Arakel E, Schwappach B, et al (2016) The molecular and functional identities of atrial cardiomyocytes in health and disease. Biochim Biophys Acta. 2016; 1863(7):1882-1893.
Vassalle, K Greenspan, S Jomain, et al. Effects of potassium on automaticity and conduction of canine hearts. Am J Physiol 1964; 207:334-340.
Parham WA, Mehdiard AA, Biermann KM, et al. Hyperkalaemia revisited. Tex Heart Inst J. 2006; 33(1): 40-47.
De Mello CW, Hoffman BF. Potassium ions and electrical activity of specialized cardiac fibers. Am J Physiol. 1960; 199: 1125-1130.
Cohen HC, Gozo EG, Pick A. The nature and type of arrhythmias in acute experimental hyperkalemia in the intact dog. Am Heart J. 1971; 82:777–785.
McLean S. et al (2000) Lidocaine-induced conduction disturbance in patients with systemic hyperkalemia. Ann Emerg Med. 2000 36, 615-618.
Norman BC, Cote E, Barrett KA (2006) Wide-complex tachycardia associated with severe hyperkalaemia in three cats J Feline Med Surg. 2006; 8(6): 372-378.
Rubanick JV, Fries Rm Waugh C, et al (2016) Severe hyperkalaemia presenting with wide-complex tachcyardia in a puppy with acute kidney injury secondary to leptospirosis. J Vet Emerg Crit Care. 2016, 26(6):858-863.
Brady WJ, Skiles J (1999) Wide complex tachycardia: ECG differential diagnosis, Am J Emerg Med 17;376.
Campbell TJ, Eyse KR, Hemsworth PD. Effects of hyperkalemia, acidosis, and hypoxia on the depression of maximum rate of depolarization by class I antiarrhythmic drugs in guinea pig myocardium: differential actions of class Ib and Ic agents. J Cardiovasc Pharmacol. 1991; 18:51-59.
Song SY, Shin HD, Seo KC et al. Hyperkalemic cardiac arrest triggered by intravenous lidocaine following axillary brachial plexus block for the creation of an arteriovenous fistula: A case report. Korean J. Anaesthesiol. 2008;55(6):756-760.
One thought on “Wide complex tachycardia – More complex than first thought?”